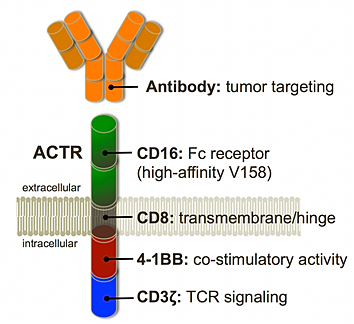
Kite Pharma had their Investor Day this week (you can watch here). I actually thought there were a number of interesting presentations and discussions that might be worth mentioning & I'll give my thoughts on them more below.
I've skipped around in the beginning, which is mostly introduction and review of all the current data.
Pipeline: Slide 9 & 10, description at 15:00 ***Slide numbers are based on those in the webcast

They gave the following timeline for their pre-IND products:
HPV E7 entering clinical trials later this year at NCI
HPV E6 entering clinical trials 2016 as a Kite IND
1st Amgen collaboration CAR product 2nd half 2016 IND
I've previously written about Kite's neoantigen TCR program, and there were some interesting developments that were mentioned at the end of the investor day - see Steven Rosenberg section below.
KRAS TCR
Additionally, I had not heard of their KRAS TCR product, which sounds very interesting. They stated the KRAS TCR program is entering clinical trials later this year at NCI, quite soon. People have tried to find ways to target "undruggable" but very commonly mutated genes in cancer like KRAS by trying to generate a T cell response (vaccines etc.) against the mutated portion of the protein, but with little success so far. If one could generate an effective response, it would be very exciting, as the mutations are clearly cancer driver mutations, present in a large percentage of tumors, and importantly also relatively homogeneous within the tumor, and would be difficult for the cancer to evolve around. So, while the idea is great if you had an effective KRAS mutant-specific TCR, the attempts so far have been underwhelming, so why would it be different here, and how did they develop this TCR?
A clue may have come later in the presentation by Dr. Steven Rosenberg of NCI, presenting on identification of neoantigen-reactive TCRs, which I'll get back to later, put up this slide (slide 96, 1:39:20)

While identifying neoantigen-reactive TCRs, they found one that was reactive against the common KRAS G12D mutation. G12D is extremely prevalent in a number of tumors:

From fantastic Ras review by McCormick and colleagues, Open Archive Here
So, it appears that by chance, they've discovered a TCR that is reactive against G12D, and like all TCRs, it is restricted against a specific HLA allele, HLA-A11, disclosed here (HT @SkepticalPhD). I am assuming that this naturally occurring TCR is the one that they are using in their upcoming clinical trial later this year, but that does not appear to have been disclosed yet. I expect since Dr. Rosenberg presented this currently unpublished data that we will be seeing a publication relatively soon on all of this. Also, if they continue to catalog what are the neoantigens and identify TCRs from TILs (tumor-infiltrating lymphocytes) that can react against them, they may be able to build up a bigger library of TCRs against shared driver mutations across a variety of HLA alleles. For me, this was the most exciting development coming out of Kite's presentation, and I'll be following the KRAS TCR story closely. There were a number of other interesting topics discussed and developments.
Multiple CAR Inputs - Inhibitory CARs
CARs are probably the most sensitive of antibody-targeted approaches for killing cancer cells. This has obvious advantages, but the disadvantage is that you need an exquisitely specific target that is not present at all (or certainly not at significant levels) on essential normal tissue. This severely limits the number of targets that make sense for CARs to be designed against. Recently a number of groups, including the Sadelain lab (Juno IP), have designed CARs that incorporate multiple inputs to improve their specificity. Essentially you are adding logic gates to the T cell so that it is designed to target cells that have:
Target A AND Target B: http://www.ncbi.nlm.nih.gov/pubmed/23242161
Target A NOT Target B: http://www.ncbi.nlm.nih.gov/pubmed/24337479
Target A OR Target B: http://www.ncbi.nlm.nih.gov/pubmed/23839099
While the first two examples were from the Sadelain lab, other groups are working on building similar systems, such as Martin Pule (IP I believe licensed to Autolus). Other labs seem to be working on additional approaches as well.
On Slide 43 (53:55) Kite revealed plans for an inhibitory CAR (the NOT approach above) to improve on-target off-tumor specificity & potentially open up new targets. The presenter said they could not reveal the details yet as this is still ongoing work, but it will be interesting to see what targets they go after if these make it to the clinic at Kite, Juno, or other CAR players.

Screening for TCRs
Ton Schumacher, now CSO at Kite EU, presented on technology he has developed to screen, similar to how antibodies can be screened, for TCR binding to a given target (starting at slide 48, 1:00:00).


In addition to screen for TCRs with optimal properties, it is possible that this approach could help overcome one of the drawbacks of TCR-based therapies, which is that they have to be specific to a patient's HLA allele, and any given TCR will not be able to be given to every patient. This screening technology was specifically mentioned as a way to generate TCRs against different HLA-alleles for HPV antigens to be able to cover about 90% of patients (at 2:17:00). Schumacher's group has also developed ways of screening patient TCRs for tumor reactivity, a potential approach for developing patient-specific neoantigen TCRs.
Steven Rosenberg
Rosenberg presented starting at slide 56, 1:20:40, and even though he was summarizing previous results, I thought the whole thing was worth a listen.
I'll just quickly mention some interesting slides he presented on upcoming CAR & TCR programs first.
I'll just quickly mention some interesting slides he presented on upcoming CAR & TCR programs first.
Slide 79, 1:31:50
He mentioned a Thyroglobulin CAR for thyroid cancers entering clinical trials at NCI. I thought this was interesting as it's a tissue-specific target, that might be the most similar to CD19 in terms of a target present on a non-essential tissue that you it is acceptable to ablate (the thyroid here, vs. B cells with CD19). I would be interested in other targets present on non-essential tissues as CAR targets, perhaps others will still need added specificity of multiple CAR inputs, as previously mentioned above.
Slide 82, 1:34:00
When discussing targeting Cancer/Testes (CT) antigens, he mentioned that while in certain tumor types NY-ESO-1 is re-expressed frequently, across all epithelial tumors, only 1.8% express NY-ESO-1 on >50% of their cells. NY-ESO-1 directed TCRs have already shown efficacy in synovial sarcoma and melanoma.
MAGE A3 on the other hand is present in 17.8% of epithelial tumors on >50% of cells. They are now concentrating making better TCRs here against MAGE A3, as numerous, and fatal cross-reactivity had occurred with previous TCRs against this target, describe here, here and here.
If they can develop a safe & effective TCR against MAGE-A3 it could b
If they can develop a safe & effective TCR against MAGE-A3 it could b
Slide 95, 1:38:30 & Slide 96
Now getting to patient-specific neoantigen TCRs, Rosenberg pointed out the potential benefit of inserting TCRs into T cells as opposed to using TILs directly, specifically that you can put the TCR into the right T cell, not an exhausted, or more differentiated T cell that might be more prevalent in TIL therapies. TILs have already had some remarkable activity, specifically in melanoma, and additionally Rosenberg has recently published a case report in Science, where they purified a specific T cell clone reactive against a patient-specific neoantigen and expanded those to make a new T cell product given to the patient with impressive efficacy. While this first patient, presumably patient 1 in the table below, was treated with purified TILs, Rosenberg's group has gone on to identify the neoantigens the TILs can react against as well as identify the TCRs that are reacting against them.
I am not sure if the patients listed above were just analyzed to see if they could identify TCRs against patient-specific mutations, or if they were treated with either a specific TIL clone reactive against the antigen or with a neoantigen-specific TCR. It will certainly be interesting to see what comes next with these approaches that presumably will be coming relatively soon in publications or presentations.
Manufacturing of an autologous T cell product reactive against a patient-specific mutation seems like a daunting task, but, perhaps it's not quite as daunting as I previously thought. In the Q&A at 2:01:30 - Rosenberg stated that it only takes 48 hours to sequence the exome from a tumor, and then only an additional 48 hours to identify which peptides potentially bind the patient's MHC. So maybe with approaches Rosenberg has described in reviews here, or those previously mentioned above developed by Ton Schumacher, the development of patient-specific neoantigen-reactive TCRs will be within the realm of feasibility.
T Cell Product Composition
T Cell Product Composition
Another
interesting point that is not mentioned very often, but probably
influences the quality & persistence of all T cell products, whether
CAR or TCR, is the differentiation status of the T cells in the cell
product before administration. I don't think it's completely understood
what's the ideal mix of the different levels of differentiated T cells.
This topic was discussed on slides 41 & 42, starting at 49:35, for
those interested:

Researchers at the NCI have published a methods paper for generating high central-memory phenotype T cell products here, which might be what they already use: http://www.ncbi.nlm.nih.gov/pubmed/20551831

Researchers at the NCI have published a methods paper for generating high central-memory phenotype T cell products here, which might be what they already use: http://www.ncbi.nlm.nih.gov/pubmed/20551831
Kite
is looking at using a small molecule to be able to expand T cells while
maintaining a more immature differentiation state for future T cell
products (slide 42)
 Here are some papers from NCI on modulating the differentiation state of T cells:
Here are some papers from NCI on modulating the differentiation state of T cells:

Other Q&A Notes (1:41:30)
2:02:50 - Allogeneic CAR-T cells
In general there are a number of barriers to effective allogeneic CAR-T therapy.
Immune suppress patients (normally not continued indefinitely)
Rejected even with major MHC matched or removed, because of minor MHC
GvH dangerous —> Rosenberg doesn’t see allo happening now
Universal NK cells —> 10^11 NK cells that can recognize cancer cell lines in vitro & no effect in patients (Rosenberg), maybe some evidence in hematologic cancers, but in solids not aware, but maybe NK approaches can be used to improve ADCC.
2:11:45 - Persistence of T cells
Long term T cell persistence might not be necessary, but it might be just correlated with good T cells that can recognize tumor.
Appears only a few weeks needed to get CR (After 1 month after treatment in Kite study, even if persistence waned after that the CRs could still be maintained - presented at AACR15).
This might have implications for thinking about how long allogeneic T cells might need to persist for sufficient activity before they are rejected by the patient's immune system.
This might have implications for thinking about how long allogeneic T cells might need to persist for sufficient activity before they are rejected by the patient's immune system.
2:14:10 - HPV E6 Antigen - why so excited?
TIL data good
Already have responses in HPV E6 TCR program, but this is not the place to disclose the full dataset. Goals now to increase response rates, need to make HLA-specific —> Schumacher tech allows identification of HPV TCRs against multiple HLA types —> so can cover 90% of the population.
Bluebird collaboration for next-gen: http://ir.kitepharma.com/releasedetail.cfm?ReleaseID=918816
Sounds like PD-1 knockout, or something similar.
Bluebird collaboration for next-gen: http://ir.kitepharma.com/releasedetail.cfm?ReleaseID=918816
Sounds like PD-1 knockout, or something similar.
2:19:05 - Target multiple mutations simultaneously?
Ongoing CRs from TILs —> TILs recognizing 1 or more, but normally number is pretty small, just a few 1, 2, 3 & still the can avoid escape.
Would like to target multiple or target a driver mutation
Mesothelin CAR-T —> probably not a good target because on a normal tissue (although they are still doing dose escalation trial too).
Conclusions
I feel like Kite/NCI has been building a really exciting TCR platform, but TCRs seem to be getting overshadowed by the hype (somewhat deserved) around CARs. I think Kite is going after the targets that seem to make the most sense, with the highest tumor-specificity -
Cancer/Testes antigens --> such as NY-ESO-1 & MAGE A3
Viral antigens --> such as HPV E6 & E7
Neoantigens --> which in general are patient-specific, but through this approach, they've already found a TCR targeting the ubiquitous KRAS G12D mutation.
So I will be very interested in following the ongoing developments in the TCR space, particularly against neoantigens & KRAS G12D, and I expect, based on Rosenberg's presentation, that we will be seeing additional publications or presentations in the not too distant future.
Disclosure: I own shares of KITE
Conclusions
I feel like Kite/NCI has been building a really exciting TCR platform, but TCRs seem to be getting overshadowed by the hype (somewhat deserved) around CARs. I think Kite is going after the targets that seem to make the most sense, with the highest tumor-specificity -
Cancer/Testes antigens --> such as NY-ESO-1 & MAGE A3
Viral antigens --> such as HPV E6 & E7
Neoantigens --> which in general are patient-specific, but through this approach, they've already found a TCR targeting the ubiquitous KRAS G12D mutation.
So I will be very interested in following the ongoing developments in the TCR space, particularly against neoantigens & KRAS G12D, and I expect, based on Rosenberg's presentation, that we will be seeing additional publications or presentations in the not too distant future.
Disclosure: I own shares of KITE